Nuove frontiere per il trattamento della SLA: la proteina HuD, un nuovo possibile target terapeutico Lo studio, pubblicato su Nature Communications e coordinato dal Dipartimento di Biologia e biotecnologie Charles Darwin della Sapienza, ha osservato un coinvolgimento della proteina HuD nei difetti della giunzione neuromuscolare, tra i primi segni distintivi della patologia. I risultati aprono allo sviluppo di nuove terapie mirate.
La SLA è una malattia neurodegenerativa progressivamente invalidante, dovuta alla compromissione graduale dei neuroni motori, le cellule nervose che stimolano la contrazione muscolare permettendo il movimento e altre funzioni importanti. Quando nei pazienti i neuroni motori degenerano, i muscoli volontari non ricevono più stimoli dal cervello e si atrofizzano, portando così alla paralisi completa. Attualmente, si utilizzano trattamenti capaci di ridurre i sintomi della malattia ma non esiste una cura per fermarne la progressione.
I difetti della giunzione neuromuscolare – il punto di connessione tra i neuroni motori e il muscolo – sono tra i primi segni distintivi della SLA.
Da quanto emerge dalla ricerca, condotta dal gruppo di Alessandro Rosa del Dipartimento di Biologia e biotecnologie della Sapienza in collaborazione con l’Istituto Italiano di Tecnologia (IIT) e l’Università di Pittsburgh, ci sarebbe un legame tra la disregolazione di una specifica proteina, denominata HuD, e i disturbi della giunzione neuromuscolare nei pazienti affetti da SLA.
“I risultati ottenuti individuano in questa proteina un ruolo cruciale in un momento precoce della malattia suggerendola quindi come un possibile target in ambito terapeutico”, ha sottolineato Alessandro Rosa.
La ricerca ha mostrato come livelli elevati di proteina HuD possano portare a difetti alla giunzione neuromuscolare con conseguente degenerazione dei neuroni motori. Riducendo quindi i livelli della proteina con terapie mirate si potrebbero limitare i disturbi della giunzione neuromuscolare nei pazienti.
Questa evidenza è stata confermata in vivo in un modello animale, il moscerino Drosophila melanogaster, in cui la sovraespressione della proteina causa difetti nella locomozione, mentre la sua riduzione migliora il fenotipo motorio.
Il progetto è stato finanziato dal PNRR nell’ambito del Centro Nazionale 3 – Sviluppo di terapia genica e farmaci con tecnologia a RNA. Per il Dipartimento di Biologia e Biotecnologie hanno contribuito al lavoro anche Alessio Colantoni e Monica Ballarino.
Riferimenti bibliografici:
“HuD impairs neuromuscular junctions and induces apoptosis in human iPSC and Drosophila ALS models” – Beatrice Silvestri, Michela Mochi, Darilang Mawrie, Valeria de Turris, Alessio Colantoni, Beatrice Borhy, Margherita Medici, Eric Nathaniel Anderson, Maria Giovanna Garone, Christopher Patrick Zammerilla, Marco Simula, Monica Ballarino, Udai Bhan Pandey & Alessandro Rosa, Nature Communications, volume 15, Article number: 9618 (2024) doi: https://www.nature.com/articles/s41467-024-54004-8
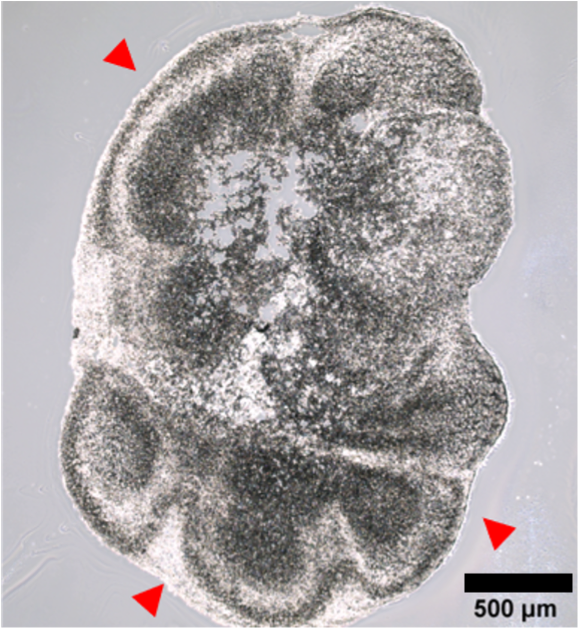
Istituto Italiano di Tecnologia – IIT e Sapienza Università di Roma: realizzati in laboratorio per la prima volta organoidi cerebrali per lo studio della Sindrome dell’X fragile Questo risultato consentirà di studiare in vitro il meccanismo molecolare della malattia e testare futuri farmaci.
Riprodotti per la prima volta in laboratorio organoidi cerebrali (3D) come modello di studio della Sindrome dell’X Fragile, una malattia ereditaria legata a mutazioni nel gene FMRP localizzato sul cromosoma X, causa di disabilità cognitiva, problemi di apprendimento e relazionali.
Lo studio, pubblicato sulla rivista Cell Death and Disease, è il risultato di una collaborazione tutta italiana fra Istituto Italiano di Tecnologia – IIT e Sapienza Università di Roma. In particolare tra Silvia Di Angelantonio e Alessandro Rosa, entrambi docenti Sapienza e ricercatori affiliati presso il centro IIT di Roma “Center for Life Nano & Neuro-Science” coordinato da Giancarlo Ruocco e il gruppo D3Validation dell’Istituto Italiano di Tecnologia di Genova, coordinato da Angelo Reggiani.
Gli organoidi 3D sono strutture cellulari tridimensionali artificiali, generate a partire da cellule staminali umane, che riproducono le caratteristiche dei veri organi. Si tratta di modelli in vitro che mostrano condizioni molto simili a quelle umane sia dal punto di vista fisiologico che patologico e che mimano in vitro l’interazione tra cellule. Negli ultimi anni la messa a punto di organoidi cerebrali umani derivati da cellule staminali pluripotenti indotte (cellule iPS, Premio Nobel per la Medicina 2012) ha permesso di ridurre i test condotti su modelli animali e ha aperto nuovi orizzonti per lo studio delle malattie del neuro-sviluppo come autismo e schizofrenia o della nota infezione da Zika virus.
Le cellule iPS sono cellule staminali che si possono ottenere ‘riprogrammando’ cellule non staminali, per esempio del sangue o della pelle, prelevate da qualunque individuo adulto.
In questo studio le colture cellulari classiche (2D) e gli organoidi cerebrali (3D) sviluppati a partire da cellule iPS, riproducono in vitro alcune caratteristiche tipiche della sindrome dell’X Fragile, consentendo ai ricercatori di studiare il meccanismo molecolare della patologia e di dimostrare come la proteina FMRP sia necessaria per supportare correttamente la proliferazione delle cellule neuronali e gliali e per impostare il corretto rapporto eccitazione-inibizione nello sviluppo del cervello umano.
Lo studio su modelli cellulari 3D, inoltre, ha permesso di scoprire uno squilibrio di dimensioni tra organoidi X fragile e organoidi di controllo cioè sani, ma soprattutto uno squilibrio in termini di bilancio eccitazione – inibizione delle cellule di X Fragile a favore dell’ipereccitabilità che si potrebbe ipotizzare essere alla base delle crisi epilettiche, sintomi tipici dei pazienti X Fragile.
Questi risultati ampliano le conoscenze sulla Sindrome dell’X Fragile e gettano le basi per lo screening di nuovi farmaci efficaci per questa patologia oltre al riposizionamento di quelli già in uso.
“Ad oggi questo lavoro è il primo a dimostrare la possibilità di studiare la Sindrome dell’X Fragile in organoidi cerebrali e suggerisce che questa piattaforma sperimentale possa essere applicata per modellizzare in vitro la Sindrome dell’X Fragile” dichiara Silvia Di Angelantonio, ricercatrice affiliata presso il centro IIT – Center for Life Nano & Neuro-Science e docente Sapienza.
“L’uso di organoidi umani per lo studio di malattie come la Sindrome dell’X Fragile presenta notevoli vantaggi per la comprensione dei meccanismi molecolari che ne sono alla base” aggiunge Alessandro Rosa, ricercatore affiliato presso il centro IIT – Center for Life Nano & Neuro-Science e docente Sapienza.
“La disponibilità di organoidi derivati da cellule umane crea i presupposti per la identificazione di farmaci migliori e, in un futuro prossimo, di terapie sempre più personalizzate sulle necessità del malato” conclude Angelo Reggiani, coordinatore del laboratorio D3Validation dell’Istituto Italiano di Tecnologia.
Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs – Carlo Brighi, Federico Salaris, Alessandro Soloperto, Federica Cordella, Silvia Ghirga, Valeria de Turris, Maria Rosito, Pier Francesca Porceddu, Chiara D’Antoni, Angelo Reggiani, Alessandro Rosa and Silvia Di Angelantonio – Cell Death & Disease https://doi.org/10.1038/s41419-021-03776-8
Testo e foto dell’Istituto Italiano di Tecnologia – IIT, Image Library; dal Settore Ufficio stampa e comunicazione Sapienza Università di Roma